SPETTROSCOPIA UV – VISIBILE
Settembre 19, 2024SPETTROSCOPIA UV – VISIBILE
Se una radiazione colpisce la materia, può essere da questa assorbita, trasmessa, riflessa, diffusa o può dare luogo a fotoluminescenza, intendendo per fotoluminescenza un certo tipo di effetti quali fluorescenza, fosforescenza ed effetto Raman.
La luce solare è composta da un insieme di radiazioni elettromagnetiche chiamate fotoni ognuno dei quali ha un’energia pari ad E=hν che si muovono nello spazio in linea retta. Ogni radiazione o fotone che compone la luce è costituito da 2 onde perpendicolari: un’onda elettrica ed una magnetica (da qui il nome di onda elettromagnetica)
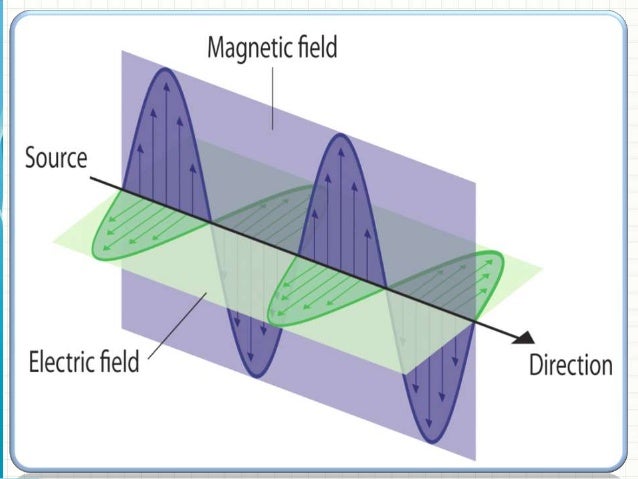
Le caratteristiche di una radiazione sono :
1) la lunghezza d’onda che si indica con λ che si misura in nanometri (nm) ed è la distanza tra due punti di massimo di 2 onde consecutive
2) una frequenza che si indica con ν ed è il numero di onde che attraversa un dato punto in 1 secondo .
3 ) l‘ampiezza cioè l’altezza dell’onda nel punto di massimo
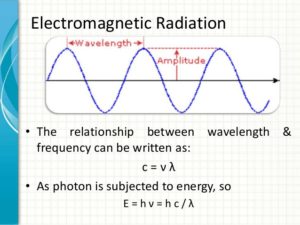
Tra λ e ν esiste la relazione :
λ = velocità/ frequenza .
Per una radiazione luminosa si ha quindi
λ = C/ ν dove C è la velocità della luce (300.000 Km /sec) quindi l’energia è E=hC/λ
Quando la luce solare colpisce un prisma si ha la scissione della luce nelle sue radiazioni che la compongono

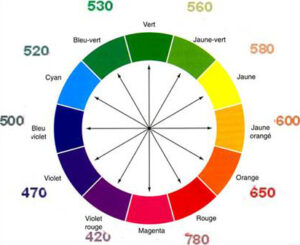
Se vediamo una soluzione di colore viola come ad es. il permanganato di potassio, la soluzione avrà assorbito il suo colore complementare che è la radiazione di colore verde.
Esiste in natura un ampio spettro di lunghezze d’onda, ma l’occhio umano riesce a vedere solo una sua piccola parte che viene chiamata regione del Visibile. L’ intero spettro di lunghezze d’onda è il seguente :
| Tipo di radiazione elettromagnetica | Frequenza | Lunghezza d’onda |
|---|---|---|
| onde radio | ≤250 MHz | 10 km – 10 cm |
| microonde | 250 MHz – 300 GHz | 1 m – 1 mm |
| infrarossi | 300 GHz – 428 THz | 1 mm – 700nm |
| visibile | 428 THz – 749 THz | 700 nm – 400 nm |
| ultravioletto | 749 THz – 30 PHz | 400 nm – 10 nm |
| raggi X | 30 PHz – 300 EHz | 10 nm – 1 pm |
| raggi gamma | ≥300 EHz | ≤1 pm |
In chimica siamo interessati soprattutto alla radiazioni UV, del Visibile ed Infrarosse che ci permettono di effettuare l’analisi quali-quantitativa dei composti e di individuare i gruppi atomici presenti nelle molecole.
L‘interazione delle molecole con le radiazioni UV e Visibile causa transizioni degli elettroni nei vari livelli, mentre nell’infrarosso vengono interessati i livelli vibro-rotazionali delle molecole.
SPETTROSCOPIA UV-VISIBILE
Quando una molecola viene colpita da una radiazione di lunghezza d’onda tra 200-400 nm (UV) o 400-800 nm (Visibile) si determinano transizioni elettroniche tra livelli energetici e ciò significa che un elettrone si muove da un orbitale molecolare di livello energetico più basso ad uno di livello energetico più alto. Come esempio semplice cosideriamo la molecola di H2 i cui Orbitali molecolari sono un σ MO di legame ed un OM σ* di antilegame. Nello stato fondamentale cioè non eccitato, i due elettroni occupano l’OM di legame con più bassa energia che viene chiamato Orbitale Molecolare più altamente occupato cioè Highest Occupied Molecular Orbital (HOMO) mentre l’OM di antilegame è definito Orbitale Molecolare meno occupato Lowest Unoccupied Molecular Orbital (LUMO).

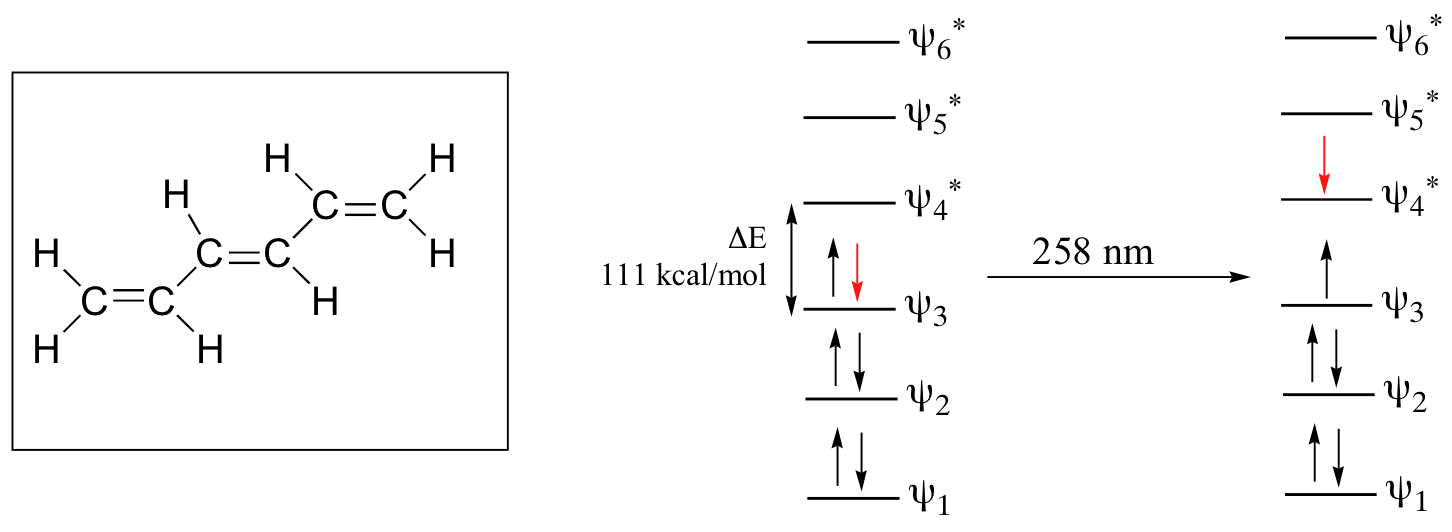
se la molecola è sottoposta all’azione di una’onda di λ con energia pari a ΔΕ cioè la differenza di energia tra i livelli LUMO ed HOMO, essa sarà assorbita e provocherà un salto di livello di un elettrone da HOMO ad LOMO cioè si verifica una transizione a σ – σ* che corrisponde all’energia di una λ = 111 nm e che in calorie è 258 Kcal/mol
Se consideriamo composti che presentano doppi legami come per esempio l’etene,

si oserva una transizione π – π* e poichè questa transizione ha un’energia più bassa della transizione σ – σ* il doppio legame assorbe a 195 nm . Queste transizioni elettroniche sono troppo energetiche per essere registrate da uno spettrofotometro UV il cui range è 200-700 nm , ma la spettroscopia UV è utilizzata per studiare le molecole organiche che hanno sistemi coniugati π – π* che assorbono a lunghezze d’onda maggiori rispetto al semplice doppio legame. Per esempio, il sistema più semplice con 2 doppi legami (Diene) è l’ 1,3-butadiene il cui OM è :
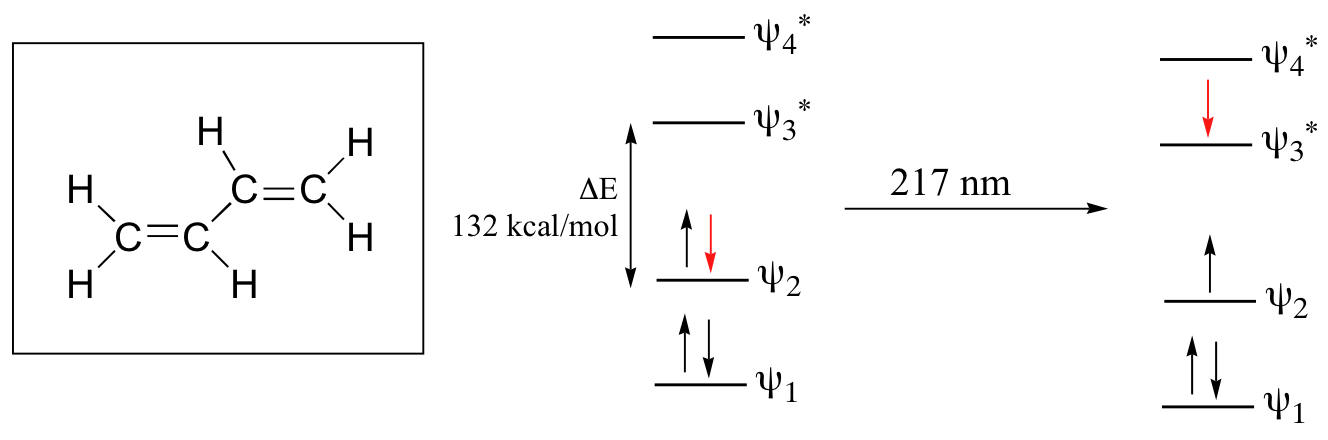
vi sono 4 orbitali π che risultano dalla combinazione di 4 orbitali atomici 2Pz. Dei 4 OM π due sono di legame e due di antilegame. Come si può vedere il ΔΕ in questo caso è più piccolo e quindi corrisponde ad una λ più grande infatti il butadiene assorbe a 217 nm. che è più grande di un solo doppio legame che è 195 nm. Più doppi legami vi sono in una molecola più piccola sarà ΔΕ relativa al salto π – π*e più grande sarà λ. Infatti
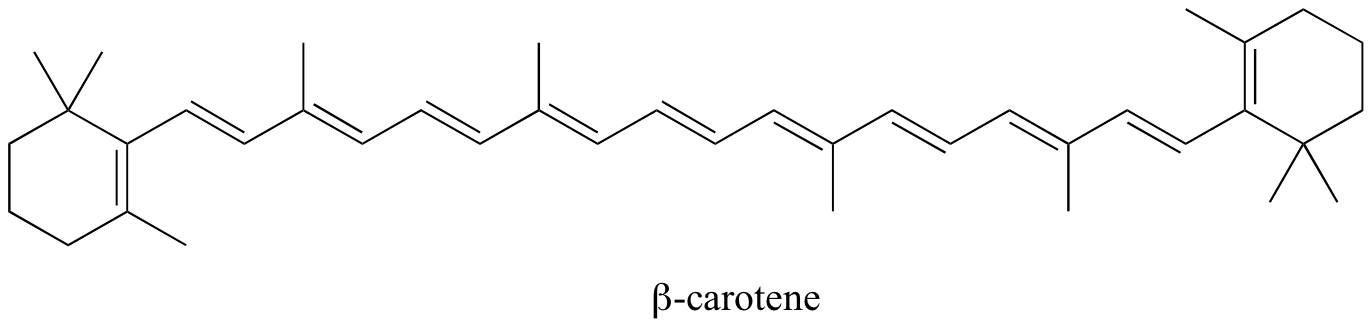
Nelle molecole con sistemi coniugati più estesi come ad es. il beta-carotene,( che ha 11 doppi legami) il ΔΕ diviene così piccolo che l’assorbimento avviene nel visibile piuttosto che nell’UV; infatti, assorbe nella regione del Blu e permette alla regione rosso-gialla di essere trasmessa (vedi ad es. il colore delle carote).
In sistemi coniugati come il 4-methyl-3-penten-2-one assorbe a 236 nm dovuto al salto π – π* ma presenta anche un assorimento a 314 nm dovuto ad una transizione di un elettrone di non legame (lone pair) dell’ossigeno del CO ad un OM π* di antilegame.
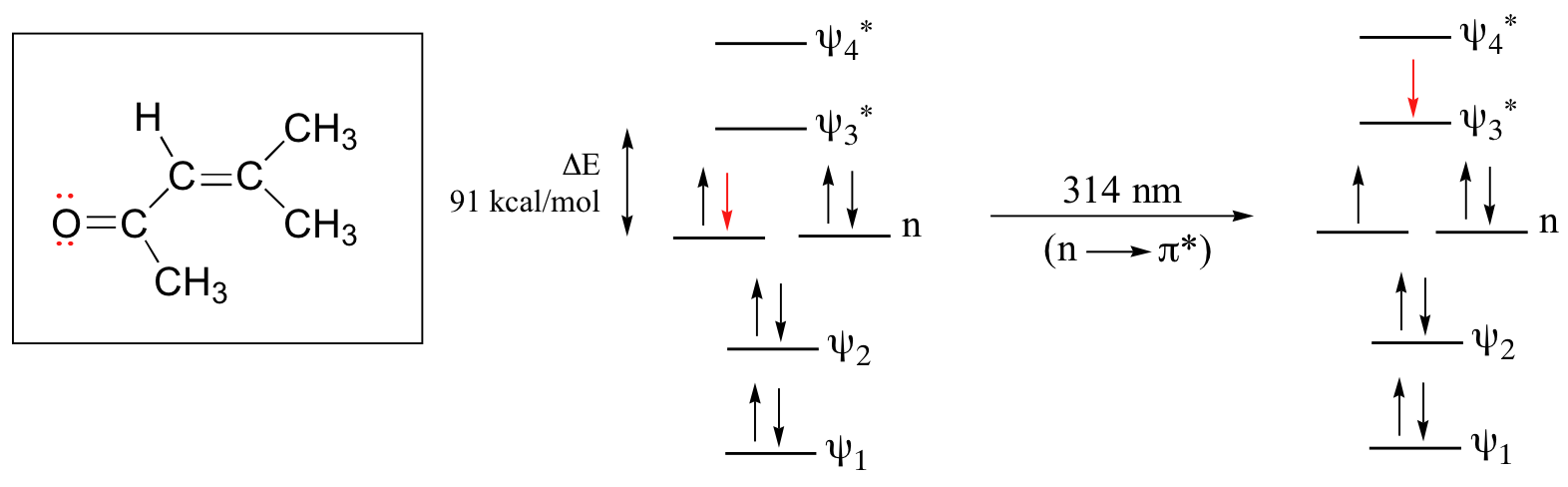
questa transizione viene indicata n→π* . Gli OM non bonding possiedono un’energia più alta che quella degli orbitali p non bonding cosicché il gap di energia per una transizione n→π ∗ n→π∗ è più piccolo di una transizione π – π* per cui l’assorbimento è ad una λ più lunga tuttavia in generale una transizione n – π* è più debole di una π – π* cioè assorbe meno radiazione.
Possiamo riassumere dicendo che nei composti che assorbono nell’UV-Visibile, le transizioni in ordine crescente di energia sono:
σ-σ* per gli alcani
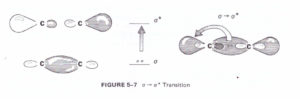
σ-π* composti carbonilici

la figura mostra le transizioni del carbonile CO
π–π* alcheni, composti carbonilici, alchini,azocomposti…
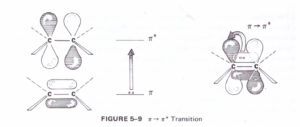
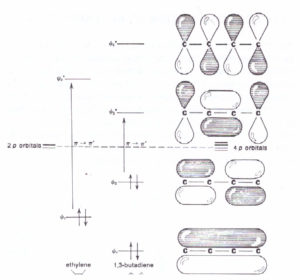
la seconda figura paragona i livelli di enrgia degli O.M. della transizione π-π* di etilene e butadiene.
π-σ* ossigeno, azoto, zolfo, composti alogenati
n-π∗ composti carbonilici (vedi la figura sopra)
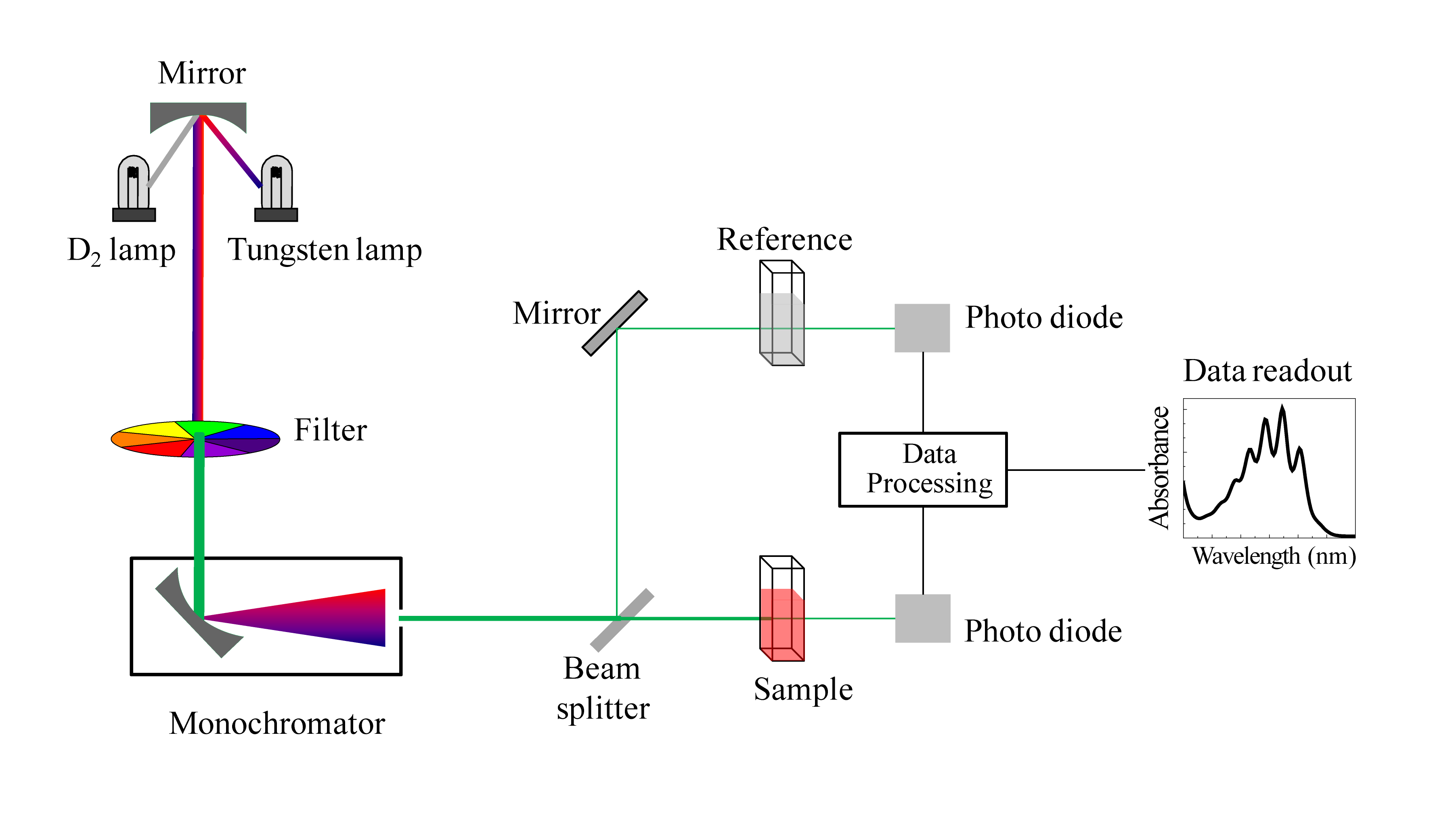
SPETTROFOTOMETRI UV-VISIBILE
Gli strumenti che si utilizzano per analizzare i composti nell’UV e nel visibile sono costituiti da :
- Una sorgente di radazioni ( diversa a seconda che sia uno strumento per UV o per visibile)
- una cella in cui è contenuto il campione
- un rivelatore
- un registratore
La lampada a tungsteno viene adoperata per ottenere lo spettro nella regione del visibile. Questa sorgente di radiazioni fornisce una radiazione continua da 320 a 3000 nm. Alla temperatura usuale di funzionamento di 3000K solo il 15 % del totale cade nella regione del visibile, e la vita media della lampada è piuttosto bassa , ma viene aumentata racchiudendo all’interno di questa lampada Iodio o Bromo a bassa pressione. Oggi si usano lampade al tungsteno con bromo o Iodio racchiuse in un’altra lampada di silice fusa (quarzo)e la lampada risultante è chiamata LAMPADA A QUARZO-ALOGENO.
Per effettuare analisi nella zona dell’ UV si usa la lampada a deuterio (o ad idrogeno) che funziona a bassa pressione e corrente DC (40 V e 5 mm di pressione del gas). Questa lampada fornisce emissione continua sino a 160 nm ma il materiale con cui è costituita la cella limita l’ operatività a 220 nm con il quarzo e 185 nm con la silice fusa.
Il meccanismo con cui è prodotto lo spettro continuo consiste nella formazione iniziale di idrogeno molecolare eccitato seguita dalla sua dissociazione per dare due specie atomiche ed un fotone ultravioletto.
H2 + Eel → H2* → H’ +H” + hν
dove Eel è l’energia elettrica assorbita dalla molecola.
L’energia totale del processo è:
Eel = EH2*= EH’ + EH” +hν
dove EH’ + EH” sono l’energia cinetica dei due atomi .
La somma di queste due energie varia continuamente da zero a EH2* quindi anche la frequenza ν del fotone prodotto, varia in modo continuo.Quindi, quando le due energie sono basse la frequenza è alta e viceversa.Ne consegue che lo spettro continuo varia da circa 160 nm sino alla regione del visibile.
Per misure superiori a 360 nm si osservano sovrapposizioni dovute all’ idrogeno per cui in questa zona vengono preferite le lampade ad incandescenza.
I monocromatori sono degli analizzatori di frequenze e possono essere a prisma od a reticolo.
Un monocromatore è un insieme costituito da fessure, lenti,specchi, finestre, prismi o reticoli che permettono di isolare una sola od un piccolissimo gruppo di lunghezze d’onda adoperate per le analisi.
I monocromatori sono degli analizzatori di frequenze e possono essere a prisma od a reticolo.
Tutti i monocromatori possiedono uno slit di ingesso ed una lente per collimare il raggio su un prisma o sul reticolo
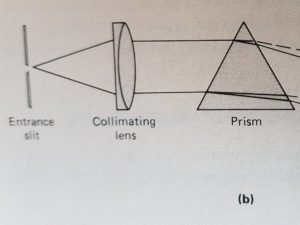
un semplice schema è mostrato in figura
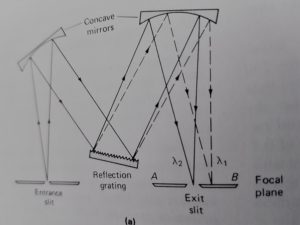
la lunghezza d’onda scelta viene convogliata su uno slit di uscita.
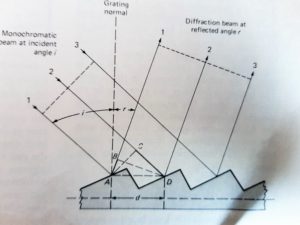
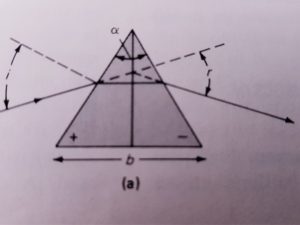
L’intervallo di lunghezze d’onda che si seleziona in uscita è determinato dall’angolo del vertice del prisma o dalla distanza delle righe del reticolo. Il prisma può essere costituito da NaCl, KBr oppure CsI.La separazione delle righe della sorgente si basa sulla rifrazione del prisma infatti il raggio entra con un angolo φ1 ed esce dal prisma con un angolo φ2 diverso dal primo a seconda dell’indice di rifrazione del prisma.
I reticoli sono di due tipi :
1- di trasmissione
2- di riflessione
Il reticolo viene costruito per stampaggio da un reticolo madre da cui si possono ottenere numerose repliche. Il reticolo madre è costruito creando un grande numero di solchi paralleli e molto vicini tra loro su una superficie levigata con uno speciale strumento che ha punte di diamante. Un reticolo per UV visibile contiene generalmente da 300 a 2000 solchi ed i più comuni sono quelli con 1200 1400 solchi.
In alcuni casi, ma solo nel visibile, vengono usati dei filtri ottici per eliminare certe frequenze, e sono di tre tipi:
1) a taglio o interdizione 2) a passa banda 3) arresta banda
I filtri ottici contengono opportune sostanze che assorbono gran parte delle radiazioni visibili lasciando solo la banda desiderata, cioè un certo intervallo di lunghezze d’onda, che ha però notevoli ampiezze (250nm). Anche combinando più filtri, rimangono comunque bande passanti dell’ordine di 50 nm e sempre a scapito di un indebolimento del raggio anche per le λ richieste. I filtri, per il loro basso costo e per le loro caratteristiche si utilizzano solo nei colorimetri.
I filtri interferenziali si basano su un fenomeno ondulatorio (l’interferenza) che causa rafforzamenti o indebolimenti tra due radiazioni che si sommano a seconda che siano o meno in fase tra loro. Sono più efficienti dei filtri basati sull’assorbimento, consentendo bande passanti dell’ampiezza di 20nm (nel visibile); sono tuttavia più costosi e si utilizzano nei colorimetri migliori.
Il raggio in uscita dal monocromatore viene splittato sia alla cella contenente il solvente sia alla cella contenente il campione disciolto nel solvente e questo elimina l’influenza del solvente e la necessità di effettuare il bianco.
Nell’ UV si utilizzano celle in quarzo (SiO2) ma nel visibile si usano celle in vetro o quarzo o in alcuni materiali plastici mentre nell’infrarosso in sono necessarie celle in NaCl, KBr, CaF2…..

Il raggio in uscita dalla cella colpisce il rivelatore di radiazioni.
I rivelatori sono dispositivi in grado di produrre un segnale elettrico che dipende dall’energia delle radiazioni che lo colpiscono. Il segnale elettrico che si genera è proporzionale all’intensità luminosa incidente e viene poi trasferito a un indicatore analogico o elaborato per via elettronica in modo più o meno complesso. I rivelatori sono una parte molto importante dello strumento sia per quanto attiene la sensibilità che per l’accuratezza dello spettrofotometro. Nell’UV si usano
1) CELLE FOTOVOLTAICHE e CELLE FOTOCONDUTTIVE;
Le celle fotovoltaiche e fotoconduttive sono costituite da semiconduttori che generano una d.d.p. direttamente proporzionale all’intensità della radiazione incidente. Esse sono però poco sensibili e non coprono tutto l’UV-visibile ma essendo resistenti e poco costose vengono utilizzate nei colorimetri o semplici fotometri di basso costo.
Una tipica cella a doppio strato è mostrata in figura
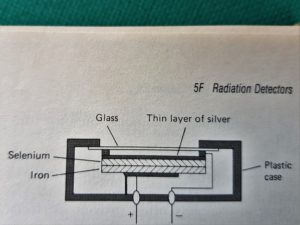
è utilizzata soprattutto per il visibile e la massima sensibilità la possiede per 550 nm. Essa consiste di uno strato di rame o ferro su cui è depositato materiale semiconduttore quale ossido di selenio la cui superficie esterna mè fissata con un film metallico trasparente di oro,argento o piombo che serve da secodo eletrrodo collettore.
Quando la radiazione colpisce il semiconduttore, i legami covalenti vengono spezzati con il risultato che si creano elettroni e buchi. Gli elettroni migrano verso il film metallico mentre i buchi la base su cui è depositatom il semiconduttore..Gli elettroni liberati possono migrare attraverso il circuito esterno per reagire con i buchi. il risultato è una corrente elettrica di grandezza proporzionale ai fotoni che raggiungono la superficie del semiconduttore e le correnti sono dell’ordine di 10 -100 microamperes.
2) FOTOTUBI e FOTOMOLTIPLICATORI;
I fototubi e i fotomoltiplicatori sono basati sull’effetto fotoelettrico, che consiste nell’emissione di elettroni da parte di un materiale quando viene colpito da radiazioni luminose: il numero di elettroni emessi (misurabile per via elettrica) è proporzionale all’intensità della radiazione incidente.
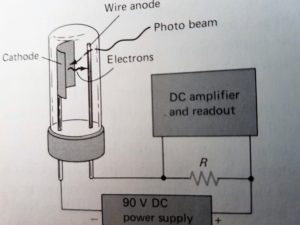

la radiazione colpisce il catodo che quindi emette elettroni captati dall’anodo e pervengono alla resistenza R detrerminando una caduta di potenziale che opportunamente amplificata raggiunge un misuratore o un registratore.
La seconda figura mostra una sezione vista dall’alto di un fotomoltiplicatore. La radiazione colpisce il catodo che così emette elettroni che poi raggiungono i cosiddetti Dinodi che sono mantenuti ad un potenziale di 9o V più positivi del catodo. Gli elettroni, nel dinodo, vengono moltiplicati e quindi per ogni fotone si producono 106-107 elettroni. Il segnale elettrico viene poi trasformato in un valore numerico proporzionale all’intensità del segnale.
3) FOTODIODI
I fotodiodi sono microscopici circuiti su chip di silicio (o germanio) che variano la loro d.d.p. se investiti da radiazioni luminose. Hanno sensibilità inferiore ai fotomoltiplicatori, ma presentano il vantaggio di poter essere inseriti in grande numero su un singolo chip di silicio, prestandosi così in modo efficace alla costruzione di spettrofotometri a serie di diodi.
SPETTROFOTOMETRI
Lo schema di uno spettrofotometro a singolo raggio è il seguente:
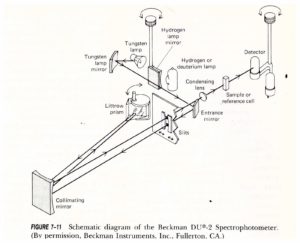
e quello a doppio raggio è:
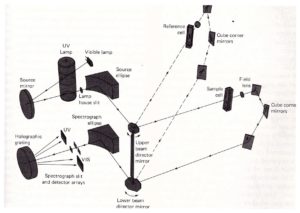
La figura seguente mostra uno spettrofotometro commerciale

Questo video ti spiega la legge che regola l’analisi quantitativa cioè la legge di Lambert e Beer
COME EFFETTUARE L’ANALISI QUALITATIVA DI UNA SOSTANZA
Per analisi qualitativa UV/Visibile intendiamo il riconoscimento di un analita all’interno di una matrice mediante scansione di uno spettro UV.
La metodica analitica prevede la preparazione del campione da analizzare sciogliendolo in un solvente , preparando quindi un bianco ed una soluzione contenente solo la matrice in modo da azzerare lo strumento. Si sottopone quindi il campione a scansione in un certo intervallo di lunghezze d’onda in cui si presume possa assorbire il nostro analita, e si osservano i picchi di massimo assorbimento. Ad ogni picco corrisponde una sostanza diversa e, siccome ad ogni picco corrisponde una lunghezza d’onda, sapendo a quale lunghezza d’onda assorbe il nostro analita oggetto di studio possiamo non solo determinare il tipo di sostanza ma possiamo procedere ad un’analisi quantitativa utilizzando la sua lunghezza d’onda di assorbimento.
La scelta del solvente è molto importante, infatti, esso dovrebbe non assorbire nell’UV nella stessa regione in cui assorbe la sostanza da analizzare.Di solito si usano solventi che non contengono doppi legami coniugati e sono perciò trasparenti alla radiazione UV.
CUT-OFF di alcuni solventi sono:
acetonitrile 190 nm n-esano 201 nm
cloroformio 240 nm metanolo 205 nm
isoottano 195 acqua 190 nm
trimetilfosfato 210 nm acetonitrile 190 nm
cicloesano 195 nm 1,4 diossano 215
etanolo 95% 205 nm
lo spettro qui sotto è lo spettro dell’isottano paragonato a quello dell’etanolo
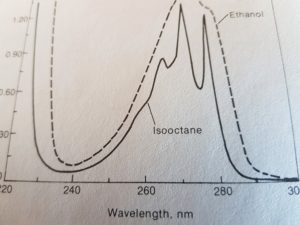
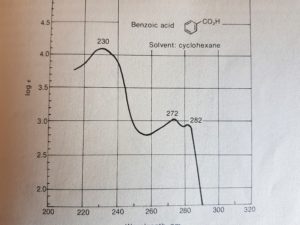
questo invecae è lo spettro dell’acido benzoico che mostra 3 picchi di assorbimento.
Di seguito è rappresentato lo spettro della nicotinamide adenine dinucleotide, abbreviato NAD+.

NAD+ presenta un picco a 260 nm e viene spesso utilizzato per alcune analisi chimico cliniche.
LA LEGGE DI LAMBERT E BEER PER L’ANALISI QUANTITATIVA SPETTROFOTOMETRICA
Abbiamo visto che quando atomi o molecole interagiscono con una radiazione incidente, gli elettroni assorbono l’energia e questo assorbimento sta alla base della legge di Lambert e Beer.
Quando una radiazione attraversa una soluzione,viene assorbita più o meno intensamente a seconda della concentrazione infatti l’assorbimento dipende dalla concentrazione della soluzione.
Se indichiamo con I0 l’ intensità della luce incidente e con I l’ intensità della luce trasmessa dalla soluzione, I sarà sempre minore di I0 perché parte è assorbita dalla soluzione, parte è riflessa, parte rifratta.
Il rapporto I/I0 viene definito trasmittanza T= I/I0
il log di I/I0 è invece definito Assorbanza A= log I/I0
A=ε Cl Legge di Lambert Beer
A=assorbanza(non ha unità dimisura)
C=concentrazione della soluzione o specie chimica in esame (moli/litro)
l=cammino ottico (cm), ovvero spessore dello strato attraversato dalla radiazione incidente
ε = coefficiente di assorbimento molare, caratteristico della sostanza (mol-1L cm-1)
Il coefficiente di estinzione molare (ε) rappresenta l’Assorbanza (o Densità Ottica) di una soluzione con concentrazione 1M e cammino ottico unitario (1cm). E’ facile calcolare il coefficiente di estinzione, infatti, basta misurare l’assorbanza A di un campione a concentrazione 1 M in una cella di 1 cm, il valore misurato coincide con ε.
Infatti A= ε x 1 M x 1 cm =ε
la legge di Lambert e Beer tuttavia è valida se:
1) la luce incidente è monocromatica
2) l’assorbimento del solvente è trascurabile
3) all’aumentare della concentrazione non si ha un aumento dell’indice di rifrazione e quindi una maggior dispersione del raggio nell’attraversare rifrazione e quindi una maggior dispersione del raggio nell’attraversare la soluzione stessa
4) non si verificano reazioni chimiche delle molecole del campione fra loro o con il solvente
Il video che segue ti spiega l’uso di uno spettrofotometro per la realizzazione di uno spettro
Questo video ti mostra come preparare un campione per l’analisi allo spettrofotometro
Non saranno presenti le bande di assorbimento alle λ di 232 nm e di 270 nm, in quanto sono assenti sistemi coniugati
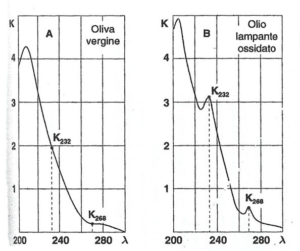
Le figure rappresentano la variazione del coefficiente di estinzione K in funzione della lunghezza d’onda tra 200 e 300 nm. La curva A è relativa agli oli vergini ed i lampanti in buono stato di conservazione. La curva B è quella tipica di un olio lampante o un olio vergine irrancidito.
MATERIALE OCCORRENTE
Strumentazione
Spettrofotometro per misure di estinzione nell’ultravioletto fra 200 e 360 nm, con possibilità di lettura per ogni unità nanometrica.
Vaschette di quarzo prismatiche (cuvette), con coperchio, di percorso ottico da 1 cm.
Le vaschette, riempite di acqua o altro solvente idoneo, non devono presentare fra di loro differenze superiori a 0,01 unità di estinzione.
Reattivi
Iso-ottano (2,2,4 trimetilpentano) puro per spettrofotometria; deve avere, in riferimento all’acqua distillata, trasmittanza non inferiore al 60 % a 220 nm e non inferiore al 95 % a 250 nm.
Vetreria
1 spruzzetta per acqua distillata
Becher da 150 mL
Matracci tarati da 25 o 50 mL
1 scatola di fazzoletti Kleenex
In un matraccio da 25 ml si pesano accuratamente ca. 0.25 g di olio o si portano a volume con isoottano. La soluzione risultante deve essere perfettamente limpida. Qualora si riscontri opalescenza o torbidità si filtra rapidamente su carta. Da questa soluzione prepararne, per pesata, una 10 volte più diluita prelevando la giusta quantità in funzione del matraccio che si ha a disposizione. Acquisizione spettro
Si azzera lo strumento con il solvente puro e si registra uno spettro UV da 200 – 340 nm. Con la soluzione dell’olio in iso-otttano si riempie una vaschetta (cuvetta) e si misurano le estinzioni, usando come riferimento il solvente impiegato, alle lunghezze d’onda significative comprese fra 232 e 276 nm. Tutti i valori letti (escluso quello a 232 nm) devono essere
compresi tra 0.1 e 0.8; viceversa si preparino soluzioni più concentrate o più diluite (vi servirà la soluzione più diluita che avete preparato).
TRATTAMENTO DEI DATI
Si calcolino i valori di estinzione specifica alle varie lunghezze d’onda in base alla K(λ) = A(λ) / (C * b) K(λ) estinzione specifica alla lunghezza d’onda λ
A(λ) assorbanza misurata alla lunghezza d’onda λ nm
C concentrazione della soluzione espressa in g / 100 ml
b spessore della cuvetta in cm I risultati si esprimono con due cifre decimali.
L’esame spettro-fotometrico dell’olio di oliva secondo il metodo ufficiale dei Regolamenti della CEE prevede la determinazione dell’estinzione specifica, in soluzione in iso-ottano, alle lunghezze d’onda di 232 e 270 nm e la determinazione del ∆K inteso come:
∆K = Km – 0.5*(Km – 4 + Km+4)
dove Km è l’estinzione specifica alla lunghezza d’onda m, lunghezza d’onda di massimo assorbimento intorno a 270nm. I parametri di K e ∆K sono parametri significativi, utili alla classificazione merceologica degli oli di oliva. NORME DI SICUREZZA
Controllate l’etichetta dell’iso-ottano e riportate sul quaderno il simbolo presente e le precauzioni che devono essere adoperate. RIFIUTI
Le soluzioni con l’olio devono essere raccolte in un unico recipiente sotto cappa. Se avete prelevato olio in eccesso non versate quanto rimane nel lavandino: raccogliete l’olio in una bottiglia che sarà buttata nel cestino dei rifiuti.
ANALISI DEI NITRATI
(da Cozzi-Protti-Ruaro “analisi chimica moderni metodi strumentali” CLUED )
Principio
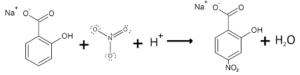
I nitrati reagiscono con sodio salicilato per formare il paranitro salicilato di sodio:
I cloruri con concentrazione superiore a 200mg/l interferiscono però possiamo eliminare l’interferenza trattando il campione con Ag2SO4 0,025 N. si filtra e si lava co H2O esente da nitrati;si riuniscono le acque di lavaggio e si procede all’analisi tenendo conto della diluizione.
Anche il Ferro interferisce in quantitrà superiori a 5 mg/l ma si può eliminare trattando il campione co ossido di Zinco agitando e filtrando.
Assieme ai nitrati tuttavia si determinano anche i nitriti se superiori a 2 mg/l . Ciò si può eliminare aggiungendo 0,05 g di (NH4)2SO4 prima dell’evaporazione(vedi sotto)
Apparecchiatura:
spettrofotometro
cuvette da 1 cm in vetro
capsule di porcellana
matracci tarat da 100 ml
pipette graduate da 10 ml
pipette tarate da 1 ml e da 50 ml
bagno di sabbia
bottiglia in polietilene
Reattivi
- Soluzione di salicilato di sodio allo 0,5010 in acqua distillata. Questa solu zione va rinnovata ogni 24 ore.
- H2S04 al 98010.
- Soluzione di NaOH e tartrato doppio di sodio e potassio.
Pesare 400 g di NaOH e 60 g di tartrato e scioglierli in un litro di acqua distilla ta in matraccio tarato.
Conservare in bottiglia di politene.
- Soluzione standard madre di nitrato.
Sciogliere 0,7220 g di KN03 (essic.cato in stufa per 2 ore a 150°C) in un litro di acqua distillata, in matraccio tarato. Aggiungere l mI di cloroformio come conservante.
Questa soluzione contiene 100,02 mg/I di azoto nitrico (ovvero 442,8 mg/I di NO3 –).
- Soluzione standard diluita di nitrato.
Prelevare 50 mI della soluzione standard madre e diluire a 500 mi con acqua di stillata, in matraccio tarato.
Questa soluzione contiene 10,002 mg/I di azoto nitrico (ovvero 44,28 mg/I di NO3-
Retta di taratura
Introdurre in una serie di capsule di porcellana da 60 mI, numerate, in successione, i reattivi come mostrato nella tab. :
| N° capsula | Bianco | l | 2 | 3 | 4 | 5 |
| Soluzione standard diluita di nitrato (ml) | O | l | 2,5 | 5 | 7,5 | 10 |
| Acqua distillata (ml) | lO | 9 | 7,5 | 5 | 2,5 | 0 |
| Salicilato di sodi o (ml) | l | l | l | l | l | l |
Si travasa il contenuto di ogni capsula in un matraccio da 100 mi e si porta a volu me con acqua distillata.Effettuate le aggiunte, si pongono le cap sule su un bagnomaria o in un bagno a sab bia, a 75-80°C. Si porta a secco facendo attenzione a non surriscaldare, nè a scal dare troppo a lungo. Si raffredda. Si ri prende il residuo con 2 mI di H2S04 conco avendo cura di bagnarlo completamente. Dopo lO minuti si aggiungono 15 mI di ac qua distillata e, cautamente, 15 mi di so luzione di NaOH e tartrato di sodi o e po tassio. A questo punte si sviluppa una co lorazione gialla.
La concentrazione in azoto nitrico di ogni soluzione staridard sarà pari a O – 0,10 – 0,25 – 0,50 – 0,75 – 1,00 mg/l, rispetti vamente.
Si procede leggendo l’assorbanza di ogni soluzione standard a 420 nm contro il bianco e riportando su un grafico i risultati ottenuti.

Determinazione del ferro nel cacao, nel cioccolato o nel caffè
(da Cozzi-Protti-Ruaro “analisi chimica moderni metodi strumentali” CLUED )
Apparecchiatura
l. Crogiolo in porcellana da 20 ml (meglio Si prelevano lO o 25 mI della soluzione clé-
se di platino). ridrica in matraccio tarato da 100 mI. Si
2. Muffola. neutralizza alla fenolftaleina con NaOH.
3. Bagno a sabbia. Si procede quindi all’aggiunta dei reattivi
4. Matracci tarati da 50 e 100 mI. come descritto per la retta di taratura. Si legge l’assorbanza a 510 nm contro un bianco trattato allo stesso modo.
Reattivi
l. HNO3 al 65%
2. HCI l: l in acqua distillata.
3. NaOH al 20% g/g
4. Fenolftaleina allo 0,1 % in alcool etilico.
Preparazione del campione . .
Pesare accuratamente 1-2 g di cioccolato spezzettato, di cacao o di caffè in un crogiolo. Procedere alla carbonizzazione su becco bunsen e trasferire in muffola a 350° . Quando le cenen sono diventate quasi bianche ( dopo 2 – 3 ore) siversano, nel crogiolo raffreddato, 1-2 ml di HNO3• Si pone il crogiolo su bagno a sabbia fino ad evaporazione dellH’NO, e infine lo si tra-sferisce di nuovo in muffola a 550°C. Le ceneri bianche vengono riprese con pochi mI di HCl 1/1 e si lascia su bagno a sabbia per almeno mezz’ora. Si raffredda e si trasferisce quantitativamente con acqua distillata in matraccio da 100 mI e si porta a volume.
Analisi dei campioni
Si prelevano 10 o 25 ml della soluzione cloridrica in matraccio tarato da 100 mI. Si neutralizza alla fenolftaleina con NaOH. Si procede quindi all’aggiunta dei reattivi come descritto per la retta di taratura. Si legge l’ assorbanza a 510 nm contro un bianco trattato allo stesso modo.
Calcoli
μg/g Fe = C· 10.000 / B·P
dove: C = μg/ml (ppm) di Fe ricavati dalla retta di taratura
p – peso di cioccolato, cacao o caffè, in grammi
B = mI di soluzione prelevati per l’analisi
